简介
文章名字: GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases
简介: 2015 年发表 于nature biotechnology, IF=54.9.作者来自马赛诸纳州,马赛诸纳州中心医院,分子病理学中心.通讯作者为J Keith Joung,来自哈弗医学院,主要从事活细胞中基因组及表观遗传重编程策略的开发,从而更好的理解生物学和疾病治疗.
研究内容
使用dsODN在RGNs进行切割时随机的NHEJ修复,通过dsODN tag特异序列进行PCR富集含有tag序列的位点,高通量测序后判断插入位点,检测脱靶发生.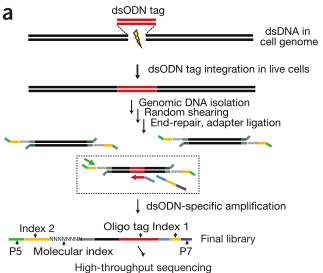
研究结果
1. GUIDE-seq方法总览
直接使用合成的dsODN效率较低,故对其进行了优化:1. 使用了5’和3’都进行磷酸化修饰的dsODN,显著提高了修复的效率;2. 开发了基于tag 序列的测序方法(图1),能够有效富集整合序列.
2. GUIDE-seq全基因组范围脱靶切割的profile
使用人的U2OS和HEK293细胞进行了验证,该部分对G-seq检测结果的基本特征进行了检测(on和off位点).
对各tag在靶点的整合位点进行了map.表示为在20bp靶序列上碱基处的reads数,以上下游的正反向引物的总的reads数表示.
各位点处均发现了与之高度相关的靶点(on 或是 off), off-target的数量呈现极大差异,和靶点无关, 和mismatch的碱基数无关, 和GC含量无关, 和分布特征无关
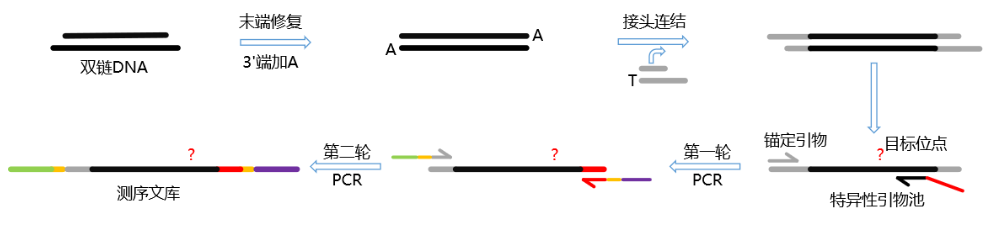
G-seq检测到的off-target处的reads和RGNs造成的不同位点的脱靶频率正相关(使用的方法检测方法为AMP测序, 测序原理如图2), 提供了一种评估剪切效率的定量方法. 同时, G-seq带有的特异性tag有效的减少了一些低频脱靶位点所需的测序深度, 表现为RGNs中未检测到的脱靶位点的检出.
3. 脱靶序列特征分析
- 对G-seq切割序列特征进行了分析: 脱靶位点的类型: 错配(高达6个碱基), 非经典PAM, 1 pb bulge-结构错配. 错配位点常发生在5’端,但在3’端(接近种子区域)也有发生, 还有off-target reads数比on-target reads数还多的情况.
- 发现:1. 随着错配碱基数增加, 脱靶位点总数减少,各脱靶位点的reads数减少, 即脱靶切割减少; 2. 影响脱靶位点数量的主要因素是错配的数量, 位点和类型.
4. 和in silico 预测结果比较
- 在检测高mismatch(4-6bp)更具优势, 计算机程序常不考虑这些高错配的脱靶位点
- 计算机预测的脱靶不够全面, 显著少于G-seq预测结果
- 计算机预测有时会忽略一些低碱基(1bp)错配的脱靶
5. 于ChIP-seq检测的binding位点比较
两者检测结果重叠率极低
6. 不依赖RGN的双链断裂的识别
只转染dsODN中,出现一些tag序列的插入. 这些位点分散在基因组中,主要集中于着丝粒和端粒, 并且可能具有细胞特异性.
7. 大片段基因组重排
AMP测序方法的应用使得染色体易位的检测成为可能. 但是,显示结果为基因组序列的直接连接, 未见tag序列. 猜测通过G-seq进行检测后,比对到基因组上, 再根据比对结果设计AMP引物进行检测,此步脱离tag-sequence.
8. 截短sgRNA的影响
证明使用截短的sgRNA显著减少了脱靶位点.
实验和表征方法
- AMP 测序; STAT-PCR:single tail adapter/tag